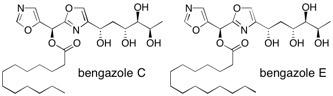
Bengazole C and E The bengazoles are marine natural products with unique structure, containing two oxazole rings flanking a single carbon. They show very potent antifungal activity. The total syntheses of bengazole C and E are described following a convergent route which involves diastereoselective cycloaddition of an appropriately substituted nitrile oxide with a butane-1,2-diacetal-protected alkenediol as the key step. See: Total Synthesis of the Potent Antifungal Agents Bengazole C and E A. Enríquez-Garcia and S.V. Ley, Coll. Czech. Chem. Comm. 2009, 74, 887-900.
2008
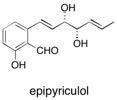
(–)-Epipyriculol A convenient synthesis of the phytotoxic natural product epipyriculol has been accomplished in 17 steps from methyl l-tartrate. The synthetic strategy is based upon the use of a butanediacetal-protected scaffold as central core from which the alkenyl side chains were assembled. See: A New Synthesis of (-)-Epipyriculol: a Phytotoxic Metabolite A. Levya, F.E. Blum and S.V. Ley, Tetrahedron, 2008, 64, 4711-4717.
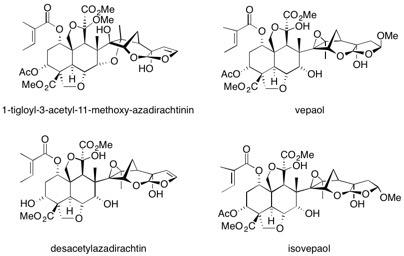
1-Tigloyl-3-acetyl-11-methoxy-azadirachtinin, vepaol, desacetylazadirachtin and isovepaol The synthesis of five natural products isolated from the Indian neem tree Azadirachta indica, is reported from a common intermediate. The judicious choice of transacetalization conditions allows efficient access to both the azadirachtinin and the azadirachtin skeletons. See: Synthesis of Natural Products from the Indian Neem Tree Azadirachta indica G.E. Veitch, A. Pinto, A. Boyer, E. Beckmann, J.C. Anderson and S.V. Ley, Org. Lett., 2008, 10, 569-572.
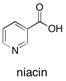
Niacin Niacin (3-picolinic acid), which is extensively used as vitamin B3 in foodstuffs and as a cholesterol-lowering agent, along with other oxygenated products of the picolines, 4-methylquinoline, and a variety of pyrimidines and pyridazines, may be produced in a single-step, environmentally benign fashion by combining single-site, open-structure, heterogeneous catalysts with a solid source of active oxygen, namely acetyl peroxyborate (APB), in the absence of an organic solvent. See: Facile, One-Step Production of Niacin (Vitamin B3) and other Nitrogen-Containing Pharmaceutical Chemicals with a Single-Site Heterogeneous Catalyst R. Raja. J.M. Thomas, M. Greenhill-Hooper, S.V. Ley, and F.A. Almeida Paz, Chem. Eu. J., 2008, 14, 2340-2348.
2007
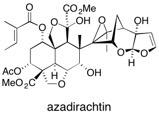
Azadirachtin We describe in full the first synthesis of the potent insect antifeedant azadirachtin through a highly convergent approach. An O-alkylation reaction is used to unite decalin ketone and propargylic mesylate fragments, after which a Claisen rearrangement constructs the central C8C14 bond in a stereoselective fashion. The allene which results from this sequence then enables a second critical carboncarbon bond forming event whereby the [3.2.1] bicyclic system, present in the natural product, is generated via a 5-exo-radical cyclisation process. Finally, using knowledge gained through our early studies into the reactivity of the natural product, a series of carefully designed steps completes the synthesis of this challenging molecule. See: Synthesis of Azadirachtin: A Long but Successful Journey G.E. Veitch, E. Beckmann, B.J. Burke, A. Boyer, S.L. Maslen and S.V. Ley, Angew. Chem. Int. Ed. 2007, 46, 7629-7632 and The Azadirachtin Story G.E. Veitch, A. Boyer and S.V. Ley, Angew. Chem. Int. Ed., 2008, 47, 9402-9429 and The Synthesis of Azadirachtin: A Potent Insect Antifeedant S.V. Ley, A. Abad-Somovila, J.C. Anderson, C. Ayats, R. Bänteli, E. Beckmann, A. Boyer, M.G. Brasca, A. Brice, H. Broughton, B.J. Burke, E. Cleator, D. Craig, A.A. Denholm, T. Durand-Reville, L.B. Gobbi, M. Gröbel, B.L. Gray, R.B. Grossmann, C.E. Gutteridge, N. Hahn, S.L. Harding, D.C. Jennens, P.J. Lovell, H.J. Lovell, M.L. de la Puente, H.C. Kolb, W-J Koot, S.L. Maslen, C.F. McCusker, A. Mattes, A.R. Pape, A. Pinto, D. Santfianos, J.S. Scott, S.C. Smith, A.Q. Somers, C.D. Spilling, F. Stelzer, P.L. Toogood, R.M. Turner, G.E. Veitch, A. Wood, C. Zumbrunn, Chem. Eur. J., 2008, 14, 10683-10704.
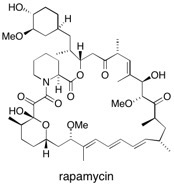
Rapamycin Rapamycin is a macrocyclic natural product, established as a potent immunosuppressant and currently of interest to the scientific community as the framework for a series of novel anticancer drugs. Extensive studies have culminated in a new convergent total synthesis of 1, which features a number of group-derived methodologies and an unusual catechol-templating strategy for the construction of the challenging macrocyclic core. See: Total Synthesis of Rapamycin M.L. Maddess, M.N. Tackett, H. Watanabe, P.E. Brennan, C.D. Spilling, J.S. Scott, D.P. Osborn and S.V. Ley, Angew. Chem., Int. Ed. 2007, 46, 591-597 and Total Synthesis of Rapamycin S.V. Ley, M.N. Tackett, M.L. Maddess, J.C. Anderson, P.E. Brennan, M.W. Cappi, J.P. Heer, C. Helgen, M. Kori, C. Kouklovsky, S.P. Marsden, J. Norman, D.P. Osborn, M.Á. Palomero, J.B.J. Pavey, C. Pinel, L.A. Robinson, J. Schnaubelt, J.S. Scott, C.D. Spilling, H. Watanabe, K.E. Wesson and M.C. Willis, Chem. Eur. J., 2009, 15, 2874-2914.
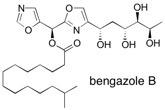
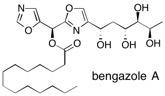
Bengazole B The bengazoles are a family of marine natural products that display potent antifungal activity and a unique structure, containing two oxazole rings flanking a single carbon atom. Total syntheses of bengazole A and B are described, which contain a sensitive stereogenic centre at this position between the two oxazoles. Additionally, the synthesis of 10-epi-bengazole A is reported. Two parallel synthetic routes were investigated, relying on construction of the 2,4-disubstituted oxazole under mild conditions and a diastereoselective 1,3-dipolar cycloaddition. Our successful route is high yielding, provides rapid access to single stereoisomers of the complex natural products and allows the synthesis of analogues for biological evaluation. See: Total Synthesis of Potent Antifungal Marine Bisoxazole Natural Products Bengazoles A and B J.A. Bull, E.P. Balskus, R.A.J. Horan, M. Langner and S.V. Ley, Chem. Eur. J. 2007, 13, 5515-5538.
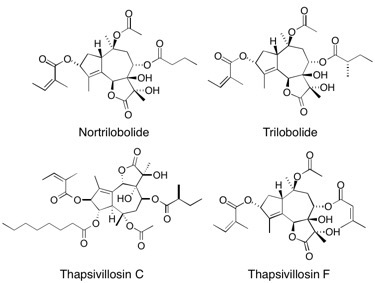
Thapsivillosin C, thapsivillosin F, trilobolide and nortrilobolide We describe the total synthesis of five guaianolide natural products: thapsigargin, thapsivillosin C, thapsivillosin F, trilobolide and nortrilobolide. Prodrug derivatives of thapsigargin have shown selective in vivo cytotoxicity against prostate tumours and the need for further investigation of this phenomenon highlights the importance of these total syntheses. The first absolute stereochemical assignment of thapsivillosin C is also delineated. See: Total Synthesis of Five Thapsigargins – Guaianolide Natural Products Exhibiting Subnanomolar SERCA Inhibition S.P. Andrews, M. Ball, F. Wierschem, E. Cleator, S. Oliver, K. Högenauer, O. Simic, A. Antonello, U. Hünger, M.D. Smith and S.V. Ley, Chem. Eur. J., 2007, 13, 5688-5712.
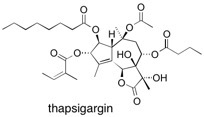
Thapsigargin The enantioselective total synthesis of thapsigargin, a potent, selective inhibitor of the Ca2+ pump SERCA, is described. Starting from ketoalcohol 8, key steps involve regioselective introduction of the internal olefin at C4−C5, judicious protecting group choice to allow chelation-controlled reduction at C3, and chemoselective introduction of the angelate ester function at C3-O. A selective esterification approach completes the total synthesis in a total of 42 steps. See: Total Synthesis of Thapsigargin, A Potent SERCA Pump Inhibitor M. Ball, S.P. Andrews, F. Wierschem, E. Cleator, M.D. Smith and S.V. Ley, Org. Lett. 2007, 9, 663-666.
2006
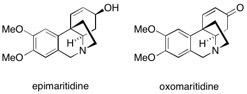
Oxomaritidine A flow process for the multi-step synthesis of the alkaloid natural product (±)-oxomaritidine is described, mediated through the use of microfluidic pumping systems that progress material through various packed columns containing immobilized reagents, catalysts, scavengers or catch and release agents; our route involves the combination of seven separate synthetic steps linked into one continuous sequence utilizing flow chemistry. See: A Flow Process for the Multi-Step Synthesis of the Alkaloid Natural Product Oxomaritidine: A New Paradigm for Molecular Assembly I.R. Baxendale, J. Deeley, C.M. Griffiths-Jones, S.V. Ley, S. Saaby and G. Tranmer, J. Chem. Soc., Chem. Commun. 2006, 2566-2568.
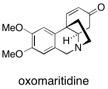
Grossamide This article describes the first enantioselective total synthesis of 2-aryl-2,3-dihydro-3-benzofurancarboxyamide neolignan, grossamide (1) using a fully automated and scalable flow reactor. See: Preparation of the Neolignan Natural Product Grossamide by a Continuous Flow Process I.R. Baxendale, C.M. Griffiths-Jones, S.V. Ley and G.K. Tranmer, Synlett 2006, 427-430.
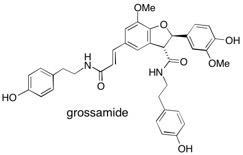
Bengazole A Candidate for Candida: A high-yielding and versatile synthesis of bengazole A has been developed; the first to provide access to a single stereoisomer of the natural product. Key steps include the construction of the 2,4-disubstituted oxazole under mild conditions and a diastereoselective 1,3-dipolar cycloaddition. See: Stereocontrolled Total Synthesis of Bengazole A: A Marine Bisoxazole Natural Product Displaying Potent Antifungal Properties J.A. Bull, E.P. Balskus, R.A.J. Horan, M. Langner and S.V. Ley, Angew. Chem. Int. Ed. 2006, 45, 6714-6718 and Total Synthesis of Potent Antifungal Marine Bisoxazole Natural Products Bengazoles A and B J.A. Bull, E.P. Balskus, R.A.J. Horan, M. Langner and S.V. Ley, Chem. Eur. J. 2007, 13, 5515-5538.
2005
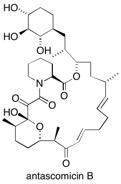
Antascomicin B The structurally enticing antascomicin B, which exhibits potent immunosuppressant-antagonizing properties, has a complex polyketide structure. Key steps in its enantioselective total synthesis include a transannular catechol-templated Dieckmann reaction to assemble the challenging tricarbonyl functionality and a butanediacetal-directed allegation. See: Total Synthesis of Antascomicin B D.E.A. Brittain, C.M. Griffiths-Jones, M.R. Linder, M.D. Smith, C. McCusker, J.S. Barlow, R. Akiyama, K. Yasuda and S.V. Ley, Angew. Chem., Int. Ed., 2005, 44, 2732-2737.
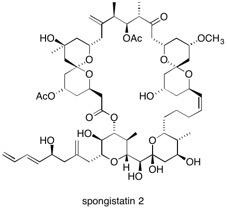
Spongistatin I The challenging structure and potent growth inhibition properties against a variety of human cancer cell lines make Spongistatin 1 (1) an exciting target for total synthesis. Enantioselective total synthesis has been achieved by exploiting a latent element of pseudo-symmetry (pseudo-C2 axis) within the ABCD bis(spiroketal) fragment (red substructure). See: Total Synthesis of Spongistatin I: A New Synthetic Strategy Exploiting its Latent Pseudo Symmetry M. Ball, M.J. Gaunt, D.F. Hook, A.S. Jessiman, S. Kawahara, P. Orsini, A. Scolaro, A.C. Talbot, H.R. Tanner, S. Yamanoi and S.V. Ley, Angew. Chem. Int. Ed., 2005, 44, 5433-5438 and Azeotropic Reflux Chromatography: An Efficient Solution to a Difficult Separation in the Scale-up of Spongistatin I M. O’Brien, A. Dieguez-Vázquez, D-S. Hsu, H. Kraus, Y. Sumino and S.V. Ley, Org. Biomol. Chem., 2008, 6, 1159-1164.

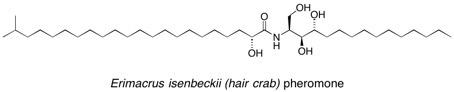
Hair crab pheromone A stereoselective synthesis of a ceramide sphingolipid as a potential sex pheromone of the hair crab Erimacrus isenbeckii is reported using diastereoselective alkylation and aldol reactions of butane-2,3-diacetal (BDA) desymmetrised glycolic acid building blocks as the key synthetic steps. See: Synthesis of a Ceramide Sphingolipid as a Potential Sex Pheromone of the Hair Crab Erimacrus isembeckii using Butane-2,3-diacetal Desymmetrised Glycolic Acid Building Blocks D.J. Dixon, S.V. Ley, S. Lohmann and T.D. Sheppard, Synlett., 2005, 481-484.
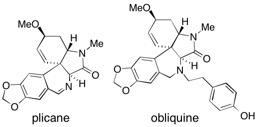
(+)-Plicane and (-)-obliquine Two new naturally occurring amaryllidaceae alkaloids have been synthesized, using a divergent approach facilitated by the use of polymer-supported reagents and scavengers. See: Synthesis of the Alkaloid Natural Products (+)-Plicane and (–)-Obliquine using Polymer Supported Reagents and Scavengers I.R. Baxendale and S.V. Ley, Ind. Eng. Chem. Res., 2005, 44, 8588-8592.
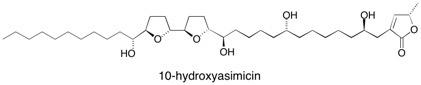
10-Hydroxyasimicin Orthogonal and modular templating is an effective basis for the preparation of the biologically active annonaceous acetogenin 10-hydroxyasimicin. The versatile tartrate-derived 2,3-butanediacetal building block together with a highly diastereoselective hetero-Diels–Alder approach to the butenolide unit were usefully employed in this novel synthetic route. See: The Total Synthesis of the Annonaceous Acetogenin, 10-Hydroxyasimicin G.L. Nattrass, E. Diez, M. McLachlan, D.J. Dixon and S.V. Ley, Angew. Chem. Int. Ed., 2005, 44, 580-584.
2004
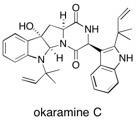
(+)-Okaramine The first total synthesis of (+)-okaramine C is described. Our previously described selenocyclisation-oxidative deselenation sequence was used to establish a 3a-hydroxy-pyrrolo[2,3- b]indole core, which was modified by selective epimerisation to the common pyrrolo[2,3- b]indole of the okaramines. See: A Concise Total Synthesis of (+) Okaramine C, E. Cleator, P.R. Hewitt and S.V. Ley, Org. Biomol. Chem., 2004, 2, 2415-2417.
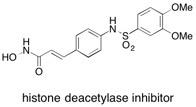
Histone Deacetylase Inhibitor The polymer-assisted solution phase synthesis (PASP) of an array of histone deacetylase (HDAc) inhibitors is described. HDAc inhibitors have considerable potential as new anti-proliferative agents. Selected compounds were shown to inhibit both human endothelial cell proliferation, and the formation of tubules (neovascularisation) in an in vitro model of angiogenesis. See: Polymer-Assisted Multistep Solution Phase Synthesis and Biological Screening of Histone Deacetylase Inhibitors A. Bapna, M. Ladlow, E. Vickerstaff, B.H. Warrington, T-P. Fan and S.V. Ley, Org. Biomol. Chem., 2004, 2, 611-620.
2003
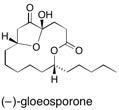
(–)-Gloeosporone The synthesis of the germination self-inhibitor (‚àí)-gloeosporone is reported. The embedded 1,7-diol motif in the product is constructed by an ironcarbonyl tether controlled Mukaiyama aldol reaction. The key step in the synthesis is the reductive removal of the ligating iron species by treatment of an acetoxycomplex 6 with lithium naphthalene. See: Synthesis of (-)-Gloeosporone, a Fungal Autoinhibitor of Spore Germination using a π-Allyltricarbonyliron Lactone Complex as a Templating Architecture for 1,7-Diol Construction E. Cleator, J. Harter, C.J. Hollowood and S.V. Ley, Org. Biomol. Chem., 2003, 1, 3263 and The Synthesis of (-)-Gloeosporone, a Potent Fungal Autoinhibitor of Spore Germination using a pi-Allytricabonyl iron Lactone Complex and a New Reductive Decomplexation Procedure for the Installation of the Embedded 1,7-Diol Component of the Natural Product E. Cleator, J. Harter and S.V. Ley, Heterocycles, 2004, 62, 619-633.
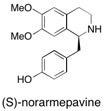
(–)-Norarmepavine We describe, in full, the enantioselective synthesis of the tetrahydrobenzylisoquinoline alkaloid (-)-norarmepavine (1) in 77% e.e. This compound was prepared using solid-supported reagents and scavengers in multi-step sequences of reactions to give materials that required no conventional purification at the individual steps. See: Enantioselective Synthesis of the Tetrahydrobenzylisoquinoline Alkaloid (-)-Norarmepavine using Polymer-Supported Reagents I.R. Baxendale, T.D. Davidson S.V. Ley and R.H. Perni, Heterocycles, 2003, 60, 2707-2715.
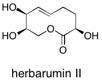
Herbarumin II The total synthesis of phytotoxic nonenolide herbarumin II has been achieved by implementation of butane diacetal (BDA)-desymmetrised glycolate building blocks. Three of the four stereogenic centres present in the key coupling fragments were generated from both enantiomeric forms of the BDA building block in highly diastereoselective alkylation and aldol reactions. See: The Use of Butane Diacetals of Glycolic Acid as Precursors for the Synthesis of the Phytotoxic Camodulin Inhibitor Herbarumin II E. Diez, D.J. Dixon, S.V. Ley, A. Polara and F. Rodríguez, Helv.Chim. Acta., 2003, 86, 3717-3729 and Total Synthesis of the Phytotoxic Agent Herbarumin II using Butane Diacetals of Glycolic Acid as Building Blocks E. Diez, D.J. Dixon, S.V. Ley, A. Polara and F. Rodriguez, Synlett., 2003, 1186.
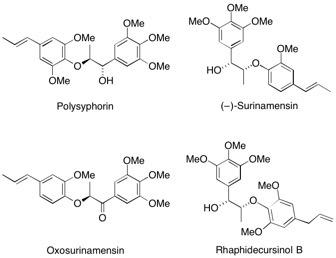
Polysphorin, oxosurinamensin, rhaphidecursinol B and (–)-surinamensin A general asymmetric route to both enantiomers of polysphorin has been developed. The route utilizes polymer-supported reagents, catalysts and scavengers to minimise the need for aqueous work-up and chromatography. This includes application of a method to scavenge 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and a “catch-and-release” procedure to extract the resultant diol following Sharpless asymmetric dihydroxylation. A novel enzymatic selective protection and investigations of a new asymmetric dihydroxylation using microencapsulated osmium tetroxide were also investigated during the course of this study. See: The Synthesis of the Anti-Malarial Natural Product Polysphorin and Analogues using Polymer-Supported Reagents and Scavengers A-L. Lee and S.V. Ley, Org. Biomol. Chem., 2003, 1, 3957.
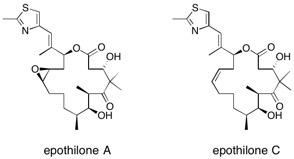
Epothilone C and A A total synthesis of epothilone C with concomitant formal synthesis of epothilone A is described, using immobilized reagents and scavengers to effect multistep synthetic transformations and purifications. See: A Total Synthesis of Epothilones using Solid-Supported Reagents and Scavengers R.I. Storer, T. Takemoto, P.S. Jackson and S.V. Ley, Angew. Chem. Int. Edn., 2003, 42, 2521 and Multi-step Application of Immobilized Reagents and Scavengers: A Total Synthesis of Epothilone C R.I. Storer, T. Takemoto, P.S. Jackson, D.S. Brown, I.R. Baxendale and S.V. Ley, Chem. Eur. J., 2004, 10, 2529-2547.
2002
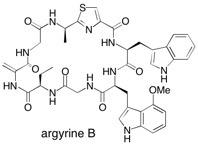
Argyrine B The total synthesis of Argyrin B is presented using a synthetic plan that is convergent and flexible and conserves the stereogenic centers. The unusual amino acid 4-methoxy tryptophan was obtained via an enzymatic resolution. Cyclization followed by oxidative elimination of the phenylseleno cysteine to the sensitive dehydroalanine afforded synthetic Argyrin B. See: Total Synthesis of the Cyclic Peptide Argyrine B S.V. Ley and A. Priour, Eur. J. Org. Chem., 2002, 3995 and Total Synthesis of the Cyclic Heptapeptide Argyrin B: A New Potent Inhibitor of T-Cell Independent Antibody Formation S.V. Ley, A. Priour and C. Heusser, Org. Lett., 2002, 4, 711.
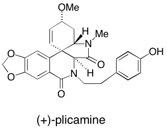
(+)-Plicamine In this report we describe in full the total synthesis of the amaryllidaceae alkaloid (+)-plicamine 1 including a model compound study. In both cases the compounds were prepared using solid-supported reagents and scavengers in multi-step sequences of reactions to give materials which required no conventional purification but could be carried on to the next synthetic step. See: Total Synthesis of the Amaryllidaceae Alkaloid (+)-Plicamine and its Unnatural Enantiomer by using Solid-Supported Reagents and Scaveners in a Multistep sequence of Reactions I.R. Baxendale, S.V. Ley and C. Piutti, Angew. Chem. Int. Edn., 2002, 41, 2194 and Total Synthesis of the Amaryllidacea Alkaloid (+)-Plicamine using Solid-Supported Reagents I.R. Baxendale, S.V. Ley, C. Piutti and M. Nesi, Tetrahedron, 2002, 58, 6285.
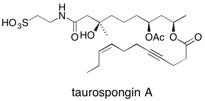
Taurospongin A The total synthesis of taurospongin A by two new approaches has been achieved where pi-allyltricarbonyliron lactone complexes have been used to control highly stereoselective additions of the nucleophiles to a carbonyl unit located in the side chain of these complexes. See: Synthesis of Taurospongin A: A Potent Inhibitor of DNA Polymerase and HIV Reverse Transcriptase using p-Allyltricarbonyliron Lactone Complexes C.J. Hollowood, S.V. Ley and S.Yamanoi, J. Chem. Soc., Chem. Common. 2002, 1624 and Use of π-Allyltricarbonyliron Lactone Complexes in the Synthesis of Taurospongin A: A Potent Inhibitor of DNA Polymerase β and HIV Reverse Transcriptase C.J. Hollowood, S.V. Ley and S. Yamanoi, Org. Biomol. Chem. 2003, 1, 1664.
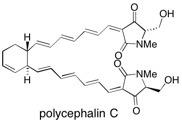
Polycephalin C The total synthesis of the polyenoyltetramic acid polycephalin C is described. Key steps of the synthesis include a double Swern oxidation, double Takai reaction and a double Stille reaction. In addition, the absolute stereochemistry of the ring junction has been determined by synthesis of both isomers and comparison of their CD spectra with natural polycephalin C. See: Total Synthesis of Polycephalin C and Determination of the Absolute Configurations at the 3”, 4” Ring Junction D.A. Longbottom, A.J. Morrison, D.J. Dixon and S.V. Ley, Angew. Chem., Int. Edn., 2002, 41, 2786 and Total Synthesis of the Polyenoyltetramic Acid Polycephalin C D.A. Longbottom, A.J. Morrison, D.J. Dixon and S.V. Ley, Tetrahedron, 2003, 59, 6955.
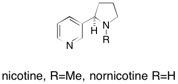
Nornicotine and nicotine The sequential use of solid-supported reagents and scavengers has led to an efficient synthesis of the natural products nornicotine, nicotine and further fuctionalised derivatives. Also reported is a diastereoselective route to both enantiomers of nicotine. See: Synthesis of Nornicotine, Nicotine and Other Functionalised Derivatives Using Solid-Supported Reagents and Scavengers I.R. Baxendale, G. Brusotti, M. Matsuoka and S.V. Ley, J. Chem. Soc., Perkin Trans. 1, 2002, 143.
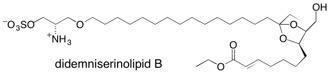
Didemniserinolipid B En route to proving the absolute and relative stereochemistry, through synthesis, of (+)-didemniserinolipid B, the first natural serinolipid isolated from a tunicate Didemnum sp., it was discovered that the isolated natural product was in fact the 31-sulfate configured 8R,9R,10R,13S,30S. This structural reassignment was only possible after the development of a microwave-assisted method for the sulfation of unreactive hydroxyl groups. See: Synthesis, Structure Revision and Absolute Configuration of Didemniserinolipid B, a Serinol Marine Natural Product from a Tunicate Didemnum sp., H. Kiyota, D.J. Dixon, C.K. Luscombe, S. Hettstedt and S.V. Ley, Org. Lett., 2002, 4, 3223.
2001
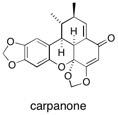
Carpanone Polymer-supported reagents have been applied to the synthesis of the natural product carpanone resulting in a clean and efficient synthesis without the requirement for conventional purification techniques. A new polymer-supported transition metal isomerisation catalyst is also reported. See: A Concise Synthesis of the Natural Product Carpanone using Solid-Supported Reagents and Scavengers I.R. Baxendale, A.-L. Lee and S.V. Ley, Synlett, 2001, 1482 and A Concise Synthesis of Carpanone using Solid-Supported Reagents and Scavengers I.R. Baxendale, A-L. Lee and S.V. Ley, J. Chem. Soc., Perkin Trans. 1, 2002, 1850.
2000
Equisetin A short stereoselective synthesis of the Fusarium toxin equisetin, a potent inhibitor of HIV-1 integrase enzyme is described, using as the key step a stereoselective intramolecular Diels–Alder reaction of a fully conjugated E,E,E-triene with a trisubstituted unsaturated β-ketothioester. See: A Short Stereoselective Total Synthesis of the Fusarium Toxin Equisetin L. T. Burke, D.J. Dixon, S.V. Ley and F. Rodríguez, Org. Lett., 2000, 2, 3611 and Total Synthesis of the Fusarium Toxin Equisetin L.T. Burke, D.J. Dixon, S.V. Ley and F. Rodríguez, Org. Biomol. Chem., 2005, 3, 274-280.

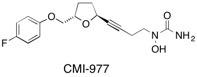
CMI-977 A short and efficient synthesis of the potent 5-lipoxygenase inhibitor CMI-977 is described using as the key step a stereoselective anomeric oxygen to carbon rearrangement of an alkynyl stannane tetrahydrofuranyl ether derivative mediated by boron trifluoride ether ate. See: A Short and Efficient, Stereoselective Synthesis of the Potent, Orally Active Anti-Histamine Agent CMI-977 D.J. Dixon, S.V. Ley, D.J. Reynolds and M. Chorgharde, Syn. Commun., 2000, 30, 1955.
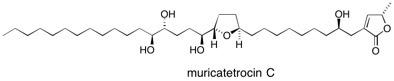
Muricatetrocin The total synthesis of the potential antitumour agent muricatetrocin C has provided an ideal stage for the exploitation and development of new chemistry. A convergent synthetic strategy has been realised incorporating three distinct pieces of methodology, these include a highly diastereoselective hetero-Diels–Alder reaction to construct the butenolide terminus, an oxygen to carbon rearrangement to install the trans-2,5-disubstituted tetrahydrofuran ring and a spatial desymmetrisation process to afford the anti-diol unit. See: The Total Synthesis of the Annonaceous Acetogenin Muricatetrocin C D.J. Dixon, S.V. Ley and D. J. Reynolds, Angew. Chem., Int. Ed. 2000, 39, 3622 and The Total Synthesis of the Annonaceous Acetogenin Muricatetrocin C, D.J. Dixon, S.V. Ley, D.J. Reynolds, Chem. Eur. J. 2002, 8, 1621.
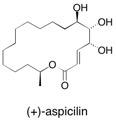
(+)-Aspicilin A short and efficient synthesis of the polyhydroxylated macrolactone (+)-aspicilin using a stereoselective lithium perchlorate mediated addition of allyltributyltin to the equatorially disposed carboxaldehyde (derived from (R‘,R‘,R,S) butane diacetal protected butane tetrol as the key step is described. Terminal group manipulation and Masamune−Roush olefination using a phosphonate ester followed by macrocyclization via ring closing metathesis afforded the natural product after partial hydrogenation and global deprotection. See: A Short and Efficient Steroselective Synthesis of the Polyhydroxylated Macrolactone (+)-Aspicilin D.J. Dixon, A.C. Foster and S.V. Ley, Org. Lett., 2000, 2, 123 and The Total Synthesis of (+)-Aspicilin using 2,3-Butane Diacetal Protected Butane Tetrols via a Chiral Memory Protocol D.J. Dixon, A.C. Foster and S.V. Ley, Can. J. Chem., 2001, 79, 1668.
α– and β-Zearalenol A highly stereoselective synthesis of the natural products α- and β-zearalenol 1 and 2 has been achieved using π-allyltricarbonyliron lactone complexes to control the 1,5-stereochemical relationship of the oxygen functionalities found in these resorcylic macrolides. See: The Use of π-Allyltricarbonyliron Lactone Complexes in the Synthesis of the Resorcylic Macrolides α- and β- Zearalenol S.V. Ley and S. Burckhardt, J. Chem. Soc., Perkin Trans. 1, 2000, 3028 and The Use of π-Allyltricarbonyliron Lactone Complexes in the Synthesis of the Resorcyclic Macrolides and Zearalenol S. Burckhardt and S.V. Ley, J. Chem. Soc., Perkin Trans. 1, 2002, 874.

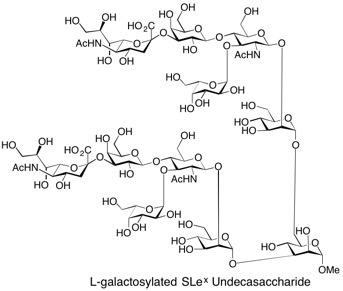
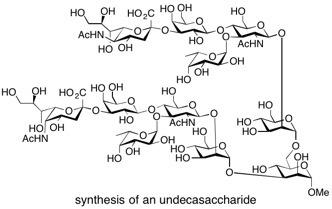
L-Galactosylated SLe(x) Undecasaccharide Galactosylated dimeric sialyl Lewis X (SLex) has been prepared employing a combination of chemical and enzymatic synthetic methods. GDP-l-galactose has been chemically synthesised. Enzymatic transfer of l-galactose onto the acceptor (Sia-α2,3-Gal-β1,4-GlcNAc-β1,3/6)2-Man-α1-OMe was achieved using the human α-1,3-fucosyltransferase V. See: Chemoenzymatic Synthesis of L-Galactosylated Dimeric Sialyl Lewis Structures Employing a-1,3-Fucosyltransferase V, A. Düffels, L.G. Green, R. Lenz, S.V. Ley, S. P. Vincent and C-H. Wong, Bioorg. Med. Chem., 2000, 8, 2519.

![chemical structure of 2,3-Dihydropyrrolo[1,2-a]quinazolin-5(1H)-one](/files/inline-images/Image_2.jpg)




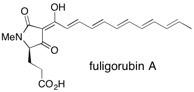
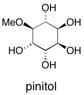
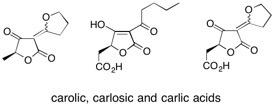
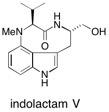

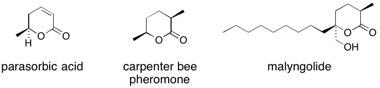

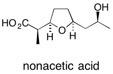
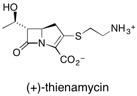

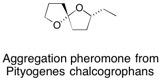
![Chemical structure of Methyl-1,6-dioxaspiro[4,5]decane](/files/inline-images/Image%2043.jpg)